- Record: found
- Abstract: found
- Article: found
Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array
Read this article at
Abstract
Background
Measurement of genome-wide DNA methylation (DNAm) has become an important avenue for investigating potential physiologically-relevant epigenetic changes. Illumina Infinium (Illumina, San Diego, CA, USA) is a commercially available microarray suite used to measure DNAm at many sites throughout the genome. However, it has been suggested that a subset of array probes may give misleading results due to issues related to probe design. To facilitate biologically significant data interpretation, we set out to enhance probe annotation of the newest Infinium array, the HumanMethylation450 BeadChip (450 k), with >485,000 probes covering 99% of Reference Sequence (RefSeq) genes (National Center for Biotechnology Information (NCBI), Bethesda, MD, USA). Annotation that was added or expanded on includes: 1) documented SNPs in the probe target, 2) probe binding specificity, 3) CpG classification of target sites and 4) gene feature classification of target sites.
Results
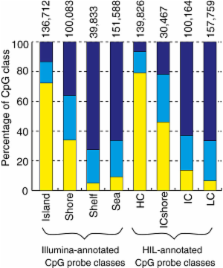
Probes with documented SNPs at the target CpG (4.3% of probes) were associated with increased within-tissue variation in DNAm. An example of a probe with a SNP at the target CpG demonstrated how sample genotype can confound the measurement of DNAm. Additionally, 8.6% of probes mapped to multiple locations in silico. Measurements from these non-specific probes likely represent a combination of DNAm from multiple genomic sites. The expanded biological annotation demonstrated that based on DNAm, grouping probes by an alternative high-density and intermediate-density CpG island classification provided a distinctive pattern of DNAm. Finally, variable enrichment for differentially methylated probes was noted across CpG classes and gene feature groups, dependant on the tissues that were compared.
Conclusion
DNAm arrays offer a high-throughput approach for which careful consideration of probe content should be utilized to better understand the biological processes affected. Probes containing SNPs and non-specific probes may affect the assessment of DNAm using the 450 k array. Additionally, probe classification by CpG enrichment classes and to a lesser extent gene feature groups resulted in distinct patterns of DNAm. Thus, we recommend that compromised probes be removed from analyses and that the genomic context of DNAm is considered in studies deciphering the biological meaning of Illumina 450 k array data.
Related collections
Most cited references24
- Record: found
- Abstract: found
- Article: not found
Principles and challenges of genomewide DNA methylation analysis.
- Record: found
- Abstract: found
- Article: found
Epigenome-wide association studies for common human diseases.
- Record: found
- Abstract: found
- Article: not found