- Record: found
- Abstract: found
- Article: found
CRISPR-Cas12a-assisted nucleic acid detection
letter
Shi-Yuan Li
1 ,
Qiu-Xiang Cheng
2 ,
Jing-Man Wang
3 ,
Xiao-Yan Li
2 ,
Zi-Long Zhang
4 ,
Song Gao
5 ,
Rui-Bing Cao
6 ,
Guo-Ping Zhao
1
,
7 ,
Jin Wang
1
,
24 April 2018
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Dear Editor,
Today, the need for time-effective and cost-effective nucleic acid detection methods
is still growing in fields such as human genotyping and pathogen detection. Using
synthetic biomolecular components, many methods have been developed for fast nucleic
acid detection
1–3
; however, they may not be able to satisfy specificity, sensitivity, speed, cost and
simplicity at the same time. Recently, a very promising CRISPR-based diagnostic (CRISPR-Dx)
(namely SHERLOCK) was established, which was based on the collateral effect of an
RNA-guided and RNA-targeting CRISPR effector, Cas13a
4
. SHERLOCK is of high sensitivity and specificity, and is very convenient in detection
of target RNA. However, to detect DNA sequences, in vitro transcription of DNA to
RNA must be conducted prior to the SHERLOCK test, which could be inconvenient.
In a recent study, we found that Cas12a, which belongs to the class 2 type V-A CRISPR-Cas
system
5
, performed collateral cleavage on non-targeted ssDNAs upon the formation of the Cas12a/crRNA/target
DNA ternary complex
6
. Here, with the employment of this feature, we used a quenched fluorescent ssDNA
reporter (e.g., HEX-N12-BHQ1 in Supplementary Table S1) as the probe, and developed
HOLMES (an one-HOur Low-cost Multipurpose highly Efficient System), which could be
used for fast detection of target DNA as well as target RNA. In HOLMES, if a target
DNA exists in the reaction system, the Cas12a/crRNA binary complex forms a ternary
complex with the target DNA, which will then trans-cleave non-targeted ssDNA reporter
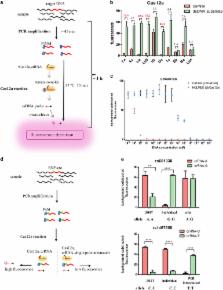
in the system, illuminating the HEX fluorescence (or any other fluorescence) (Fig. 1a).
Fig. 1
HOLMES is a rapid, simple and efficient method for nucleic acid detection.
a An illustration of HOLMES. To detect a target DNA, specific amplification of the
target DNA by either PCR or other isothermal amplification methods will be performed,
and a crRNA guide sequence is specially designed, targeting a region in the target
DNA. The PAM sequence can be designed on the primers and introduced during amplification.
After that, the amplicon was mixed with the Cas12a/crRNA complex, and a ternary complex
forms if the target DNA exists. Upon the formation of the ternary complex, the quenched
fluorescent ssDNA reporter is trans-cleaved, illuminating the fluorescence. b Comparison
of the signal-to-noise values of trans-cleavage by ten Cas12a proteins from different
species. The reaction system included Cas12a, crRNA (crRNA-T1), target DNA (pUC18-T1)
and quenched fluorescent ssDNA (HEX-N12-BHQ1), and the target DNA was omitted in the
negative control. The signal-to-noise values were labeled and values larger than 10
were shown in red (n = 3 technical replicates; bars represent the mean ± SEM). Fn
Francisella tularensis; As Acidaminococcus sp. BV3L6; Lb Lachnospiraceae bacterium
ND2006; Lb5 Lachnospiraceae bacterium NC2008; HK Helcococcus kunzii ATCC 51366; Os
Oribacterium sp. NK2B42; Ts Thiomicrospira sp. XS5; Bb Bacteroidales bacterium KA00251;
Bo Bacteroidetes oral taxon 274 str. F0058; Lb4 Lachnospiraceae bacterium MC2017.
c Detection sensitivity of Cas12a alone or Cas12a combined with PCR amplification
(i.e., HOLMES). Serially diluted pUC18-T1 plasmid was employed as the target dsDNA.
(n = 3 technical replicates; bars represent the mean ± SEM). d Schematic of human
SNP genotyping by HOLMES. The amplification of a target DNA containing the SNP locus
is almost the same as described in Fig. 1a, and design of primers and introduction
of the PAM site are detailed in Supplementary Figure S3. To detect an SNP, more than
one crRNA is needed, targeting different genotypes. e HOLMES correctly genotyped different
human SNP loci in HEK293T, a candidate individual, and the PCR-generated templates
(n = 3 technical replicates; two-tailed Student’s t-test; **p < 0.01; ****p < 0.0001;
bars represent the mean ± SEM). Genotypes verified by Sanger sequencing were annotated
below each plot, and the results of other SNP loci could be found in Supplementary
Figure S4a
We ever purified ten Cas12a proteins (Supplementary Table S3) and found all showed
the ssDNA trans-cleavage activity
6
. To find the most suitable Cas12a for HOLMES (i.e., with high signal-to-noise ratios),
we tested all ten Cas12a proteins and found Lachnospiraceae bacterium ND2006 Cas12a
(LbCas12a), Oribacterium sp. NK2B42 Cas12a (OsCas12a), Lachnospiraceae bacterium NC2008
Cas12a (Lb5Cas12a) and Francisella tularensis Cas12a (FnCas12a) showed good performance,
among which LbCas12a was chosen for the following studies (Fig. 1b). To determine
the sensitivity of HOLMES, we titrated target DNA, and found the minimum detectable
concentration for Cas12a-crRNA was approximately 0.1 nM; however, when combined with
PCR, the detectable concentration could be as low as 10 aM (Fig. 1c), which was comparable
to the SHERLOCK system
4
and was better than PCR alone or quantitative PCR using the SYBR Green method (Supplementary
Figure S1). Therefore, to achieve higher sensitivity, PCR amplification was employed
in the HOLMES test thereafter.
To test whether HOLMES could discriminate single-base differences, we made point mutations
at different positions in the target DNA sequence, including both the PAM region and
the guide sequences (Supplementary Figure S2a). When a full length of crRNA guide
sequence (24-nt crRNA, Supplementary Table S2) was used, we found mutations in either
the PAM sequences or the region of the 1st–7th bases of the guide sequence resulted
in clear decline of the fluorescence signal; however, no significant difference was
observed when the mutation was within the region of the 8th–18th bases (Supplementary
Figure S2b), which was highly consistent with the previous report that the 5′-end
seed region in the crRNA guide sequence was extremely important for Cas12a recognition
7
. In addition, based on our previous findings
8
, Cas12a with a reduced length of crRNA guide sequence showed higher cleavage specificity.
Therefore, we then tested shorter guide sequences, and found point mutations within
a larger region (1st–16th bases) resulted in more than 2-fold difference in fluorescence
signals for both 16-nt and 17-nt crRNA guide sequences (Supplementary Figure S2b),
suggesting that shorter guide sequences might be used in HOLMES. Furthermore, considering
the fact that there might exist no suitable PAM sequence nearby the SNP site, primers
for PCR amplification were specially designed to introduce the PAM sequence (Supplementary
Figure S3), which therefore allowed for sequence-independent detection of any single
nucleotide polymorphism (SNP) sites.
We then chose a dozen of SNP loci that are related to human health and personal characteristics
(Supplementary Table S4). We either extracted genomic DNA from cultured human 293T
cells or collected saliva from human individuals, and then PCR amplified the target
regions, followed by the HOLMES assay to distinguish alleles (Fig. 1d). The results
clearly showed that HOLMES had sufficiently high specificity to determine both homozygous
and heterozygous genotypes (Fig. 1e and Supplementary Figure S4a). We also collected
nineteen volunteers’ saliva samples to detect the SNP rs1014290, which is related
to gout risk, and proved that HOLMES could be used to rapidly and easily detect human
SNP genotypes (Supplementary Figure S4b).
Moreover, HOLMES could also be used to detect DNA viruses (e.g., pseudorabies virus
(PRV), Supplementary Figure S5a) and RNA viruses (e.g., Japanese encephalitis virus
(JEV), Supplementary Figure S6a), and the sensitivity for both could be as low as
1–10 aM (Supplementary Figures S5b and S6b). For JEV, total RNA was first extracted
and then reverse transcribed into cDNA before being detected by HOLMES. Because of
the high sensitivity, HOLMES successfully detected PRV virus in both the PRV-infected
cells and the culture supernatant (Supplementary Figure S5c). In addition, the high
specificity of HOLMES also enabled it to distinguish between virus strains. For example,
the PRV Ra classical strain, the cmz variant strain and the Bartha-K61 vaccine strain
were easily discriminated by the gE46 site (Supplementary Figure S5d and S5e). Similarly,
the JEV NJ2008 strain and the live-attenuated vaccine strain SA14-14-2 were well differentiated
by the site E138 (Supplementary Figure S6c and S6d).
The “SHERLOCK” nucleic acid detection system was recently established with the employment
of the “RNA collateral effect” of Cas13a and an isothermal amplification method
6
. Although both HOLMES and SHERLOCK show attomolar detection sensitivity and can be
used to detect both DNA and RNA targets, this study indicates that HOLMES may have
advantages in DNA detection, while SHERLOCK is more convenient for RNA detection.
In addition, isothermal amplification methods (e.g., the recombinase polymerase amplification
(RPA) and loop-mediated isothermal amplification (LAMP)) can also be used although
rapid PCR amplification was used in HOLMES in this study. Similar to SHERLOCK, HOLMES
requires no expensive reagents and no special instruments, making it low cost and
easily accessible for nucleic acid detection. In addition to the medical applications
described above, HOLMES may also be used for a variety of applications that require
rapid detection of nucleic acids, including monitoring foods and the environment.
(While this manuscript has been ready to submit to Cell Discovery, two pieces of work
were published on Science, both of which described the use of the Cas12a trans-cleavage
activity on ssDNAs for nucleic acid detection
9, 10
.)
Electronic supplementary material
Supplementary Information
Related collections
Most cited references2
- Record: found
- Abstract: not found
- Article: not found
Requirements for high impact diagnostics in the developing world.

- Record: found
- Abstract: found
- Article: found
The CCTL (Cpf1-assisted Cutting and Taq DNA ligase-assisted Ligation) method for efficient editing of large DNA constructs in vitro
Chao Lei, Shi-Yuan Li, Jia-Kun Liu … (2017)