- Record: found
- Abstract: found
- Article: found
Discovering Cellular Mitochondrial Heteroplasmy Heterogeneity with Single Cell RNA and ATAC Sequencing
Read this article at
Abstract
Simple Summary
Mitochondria, the powerhouse of the cell, exist in the range of 100 s–1000 s of copies in almost every cell in the body, each with their own mitochondrial DNA, called mtDNA. When the healthy operation of a significant proportion of these mitochondria is disrupted, it can lead to dysfunction and by extension disease. One source of dysfunction arises due to mutations in the mtDNA, resulting in individual cells harbouring multiple versions of mtDNA—a “standard” wild type and a variant—a state called heteroplasmy. Heteroplasmy is a state that can arise either through inheritance or by mutations that occur through life, resulting in a new mitochondrial allele within a cell. The proportion of mitochondria that have a wild type and that have a variant allele differs between individuals, tissues within an individual, and even cells within a tissue. Historically, heteroplasmy has mainly been studied with bulk sequencing technologies, which miss variation within a tissue. The cellular variation in heteroplasmy throughout the body and its implications for pathology is not fully understood. In this review article we outline recent developments in scRNA-seq and scATAC-seq techniques which allow researchers to discover the extent of this cellular variation and further uncover the role heteroplasmy plays in disease at the cellular level.
Abstract
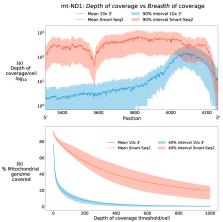
Next-generation sequencing technologies have revolutionised the study of biological systems by enabling the examination of a broad range of tissues. Its application to single-cell genomics has generated a dynamic and evolving field with a vast amount of research highlighting heterogeneity in transcriptional, genetic and epigenomic state between cells. However, compared to these aspects of cellular heterogeneity, relatively little has been gleaned from single-cell datasets regarding cellular mitochondrial heterogeneity. Single-cell sequencing techniques can provide coverage of the mitochondrial genome which allows researchers to probe heteroplasmies at the level of the single cell, and observe interactions with cellular function. In this review, we give an overview of two popular single-cell modalities—single-cell RNA sequencing and single-cell ATAC sequencing—whose throughput and widespread usage offers researchers the chance to probe heteroplasmy combined with cell state in detailed resolution across thousands of cells. After summarising these technologies in the context of mitochondrial research, we give an overview of recent methods which have used these approaches for discovering mitochondrial heterogeneity. We conclude by highlighting current limitations of these approaches and open problems for future consideration.
Related collections
Most cited references87
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.

- Record: found
- Abstract: found
- Article: found
Massively parallel digital transcriptional profiling of single cells
- Record: found
- Abstract: found
- Article: not found