- Record: found
- Abstract: found
- Article: found
The complete chloroplast genome of Amomum tsao-ko
Read this article at
Abstract
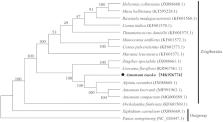
Amomum tsao-ko (Zingiberaceae) is an important edible and medicinal crop. The complete chloroplast (cp) genome of A. tsao-ko was determined using Illumina sequencing platform. The size of whole cp genome was 163,648 bp, containing a small single copy (SSC) region of 15,355 bp and a large single copy (LSC) region of 88,741 bp, which were separated by a pair of inverted repeat (IRs) regions (29,776 bp). The A. tsao-ko cp genome contained 133 genes, including eight ribosomal RNA genes (4 rRNA species), 38 transfer RNA genes (30 tRNA species) and 87 protein-coding genes (79 PCG species). The overall GC content of A. tsao-ko cp genome is 36.02%. To investigate the evolution status of A. tsao-ko, as well as Zingiberales, a phylogenetic tree with A. tsao-ko and other 16 species was constructed based on their complete chloroplast genomes. Phylogenetic analysis revealed that A. tsao-ko was closely related to Alpinia zerumbet.
Related collections
Most cited references11

- Record: found
- Abstract: found
- Article: found
MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability
- Record: found
- Abstract: found
- Article: not found
MEGA6: Molecular Evolutionary Genetics Analysis version 6.0.

- Record: found
- Abstract: found
- Article: found