- Record: found
- Abstract: found
- Article: not found
ETV1 is a lineage-specific survival factor in GIST and cooperates with KIT in oncogenesis
Read this article at

Abstract
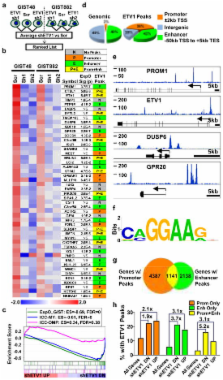
Gastrointestinal stromal tumour (GIST) is the most common human sarcoma and is primarily defined by activating mutations in the KIT or PDGFRA receptor tyrosine kinases 1, 2. KIT is highly expressed in interstitial cells of Cajal (ICCs)—the presumed cell of origin for GIST—as well as in hematopoietic stem cells, melanocytes, mast cells and germ cells 2, 3. Yet, families harbouring germline activating KIT mutations and mice with knock-in Kit mutations almost exclusively develop ICC hyperplasia and GIST 4– 7, suggesting that the cellular context is important for KIT to mediated oncogenesis. Here we show that the ETS family member ETV1 is highly expressed in the subtypes of ICCs sensitive to oncogenic KIT mediated transformation 8, and is required for their development. In addition, ETV1 is universally highly expressed in GISTs and is required for growth of imatinib-sensitive and resistant GIST cell lines. Transcriptome profiling and global analyses of ETV1-binding sites suggest that ETV1 is a master regulator of an ICC-GIST-specific transcription network mainly through enhancer binding. The ETV1 transcriptional program is further regulated by activated KIT, which prolongs ETV1 protein stability and cooperates with ETV1 to promote tumourigenesis. We propose that GIST arises from ICCs with high levels of endogenous ETV1 expression that, when coupled with an activating KIT mutation, drives an oncogenic ETS transcription program. This differs from other ETS-dependent tumours such as prostate cancer, melanoma, and Ewing sarcoma where genomic translocation or amplification drives aberrant ETS expression 9– 11 and represents a novel mechanism of oncogenic transcription factor activation.
Related collections
Most cited references17
- Record: found
- Abstract: found
- Article: not found
ChIP-seq accurately predicts tissue-specific activity of enhancers.
- Record: found
- Abstract: found
- Article: not found
PDGFRA activating mutations in gastrointestinal stromal tumors.
- Record: found
- Abstract: found
- Article: not found