- Record: found
- Abstract: found
- Article: found
Endothelial VWF is critical for the pathogenesis of vaso-occlusive episode in a mouse model of sickle cell disease
Read this article at
Significance
Current main treatments for sickle cell disease (SCD), such as hydroxyurea, L-glutamine, and crizanlizumab, reduce the frequency of vaso-occlusive episode (VOE). However, a considerable number of individuals with SCD still develop VOE. For example, although the rate was significantly reduced, VOE still occurred in 64% of individuals with SCD treated with prophylactic use of high-dose crizanlizumab, a P-selectin targeting antibody therapy newly approved by the Food and Drug Administration. Once VOE occurs, treatments are limited to supportive therapies. Thus, a better understanding of mechanisms underlying VOE pathogenesis and development of new therapies are needed to effectively manage VOE. Our study indicates that promoting von Willebrand factor cleavage by ADAMTS13 may be an effective treatment for alleviating VOE-associated organ damage.
Abstract
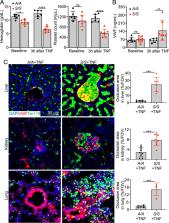
Vaso-occlusive episode (VOE) is a common and critical complication of sickle cell disease (SCD). Its pathogenesis is incompletely understood. von Willebrand factor (VWF), a multimeric plasma hemostatic protein synthesized and secreted by endothelial cells and platelets, is increased during a VOE. However, whether and how VWF contributes to the pathogenesis of VOE is not fully understood. In this study, we found increased VWF levels during tumor necrosis factor (TNF)–induced VOE in a humanized mouse model of SCD. Deletion of endothelial VWF decreased hemolysis, vascular occlusion, and organ damage caused by TNF-induced VOE in SCD mice. Moreover, administering ADAMTS13, the VWF-cleaving plasma protease, reduced plasma VWF levels, decreased inflammation and vaso-occlusion, and alleviated organ damage during VOE. These data suggest that promoting VWF cleavage via ADAMTS13 may be an effective treatment for reducing hemolysis, inflammation, and vaso-occlusion during VOE.
Related collections
Most cited references35
- Record: found
- Abstract: found
- Article: not found
Sickle-cell disease.
- Record: found
- Abstract: found
- Article: not found
Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease.
- Record: found
- Abstract: found
- Article: not found