- Record: found
- Abstract: found
- Article: not found
Modeling the mitochondrial cardiomyopathy of Barth syndrome with iPSC and heart-on-chip technologies
Read this article at

Abstract
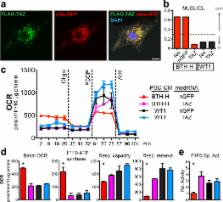
Studying monogenic mitochondrial cardiomyopathies may yield insights into mitochondrial roles in cardiac development and disease. Here, we combine patient-derived and genetically engineered iPSCs with tissue engineering to elucidate the pathophysiology underlying the cardiomyopathy of Barth syndrome (BTHS), a mitochondrial disorder caused by mutation of the gene Tafazzin (TAZ). Using BTHS iPSC-derived cardiomyocytes (iPSC-CMs), we defined metabolic, structural, and functional abnormalities associated with TAZ mutation. BTHS iPSC-CMs assembled sparse and irregular sarcomeres, and engineered BTHS “heart on chip” tissues contracted weakly. Gene replacement and genome editing demonstrated that TAZ mutation is necessary and sufficient for these phenotypes. Sarcomere assembly and myocardial contraction abnormalities occurred in the context of normal whole cell ATP levels. Excess levels of reactive oxygen species mechanistically linked TAZ mutation to impaired cardiomyocyte function. Our study provides new insights into the pathogenesis of Barth syndrome, suggests new treatment strategies, and advances iPSC-based in vitro modeling of cardiomyopathy.
Related collections
Most cited references28
- Record: found
- Abstract: not found
- Article: not found
The Tension of Metallic Films Deposited by Electrolysis
- Record: found
- Abstract: found
- Article: not found
hESC-Derived Cardiomyocytes Electrically Couple and Suppress Arrhythmias in Injured Hearts
- Record: found
- Abstract: found
- Article: not found