- Record: found
- Abstract: found
- Article: found
Cytosolic adaptation to mitochondria-induced proteostatic stress causes progressive muscle wasting

Read this article at
Summary
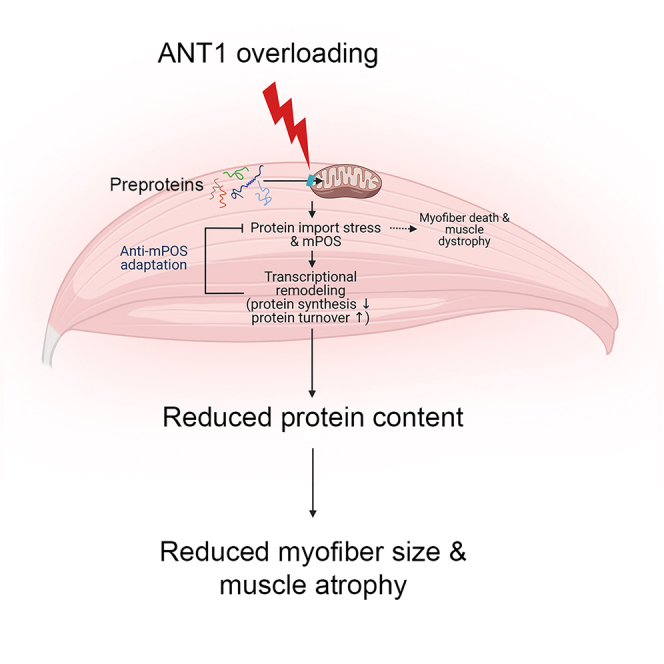
Mitochondrial dysfunction causes muscle wasting in many diseases and probably also during aging. The underlying mechanism is poorly understood. We generated transgenic mice with unbalanced mitochondrial protein loading and import, by moderately overexpressing the nuclear-encoded adenine nucleotide translocase, Ant1. We found that these mice progressively lose skeletal muscle. Ant1-overloading reduces mitochondrial respiration. Interestingly, it also induces small heat shock proteins and aggresome-like structures in the cytosol, suggesting increased proteostatic burden due to accumulation of unimported mitochondrial preproteins. The transcriptome of Ant1-transgenic muscles is drastically remodeled to counteract proteostatic stress, by repressing protein synthesis and promoting proteasomal function, autophagy, and lysosomal amplification. These proteostatic adaptations collectively reduce protein content thereby reducing myofiber size and muscle mass. Thus, muscle wasting can occur as a trade-off of adaptation to mitochondria-induced proteostatic stress. This finding could have implications for understanding the mechanism of muscle wasting, especially in diseases associated with Ant1 overexpression, including facioscapulohumeral dystrophy.
Graphical abstract
Highlights
-
•
Ant1 overexpression causes progressive muscle wasting without affecting lifespan
-
•
ANT1 overloading saturates the mitochondrial protein import pathway to cause mPOS
-
•
Muscle responds to mPOS to repress the synthesis and increase turnover of proteins
-
•
Chronic adaptation to mPOS reduces myofiber size and muscle mass as a trade-off
Abstract
Biological sciences; Cellular physiology; Cell biology; Functional aspects of cell biology
Related collections
Most cited references78

- Record: found
- Abstract: found
- Article: found
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2
- Record: found
- Abstract: found
- Article: not found
Salmon: fast and bias-aware quantification of transcript expression using dual-phase inference

- Record: found
- Abstract: found
- Article: found