- Record: found
- Abstract: found
- Article: found
Friend and foe: β-cell Ca 2+ signaling and the development of diabetes

Read this article at
Abstract
Background
The divalent cation Calcium (Ca 2+) regulates a wide range of processes in disparate cell types. Within insulin-producing β-cells, increases in cytosolic Ca 2+ directly stimulate insulin vesicle exocytosis, but also initiate multiple signaling pathways. Mediated through activation of downstream kinases and transcription factors, Ca 2+-regulated signaling pathways leverage substantial influence on a number of critical cellular processes within the β-cell. Additionally, there is evidence that prolonged activation of these same pathways is detrimental to β-cell health and may contribute to Type 2 Diabetes pathogenesis.
Scope of review
This review aims to briefly highlight canonical Ca 2+ signaling pathways in β-cells and how β-cells regulate the movement of Ca 2+ across numerous organelles and microdomains. As a main focus, this review synthesizes experimental data from in vitro and in vivo models on both the beneficial and detrimental effects of Ca 2+ signaling pathways for β-cell function and health.
Major conclusions
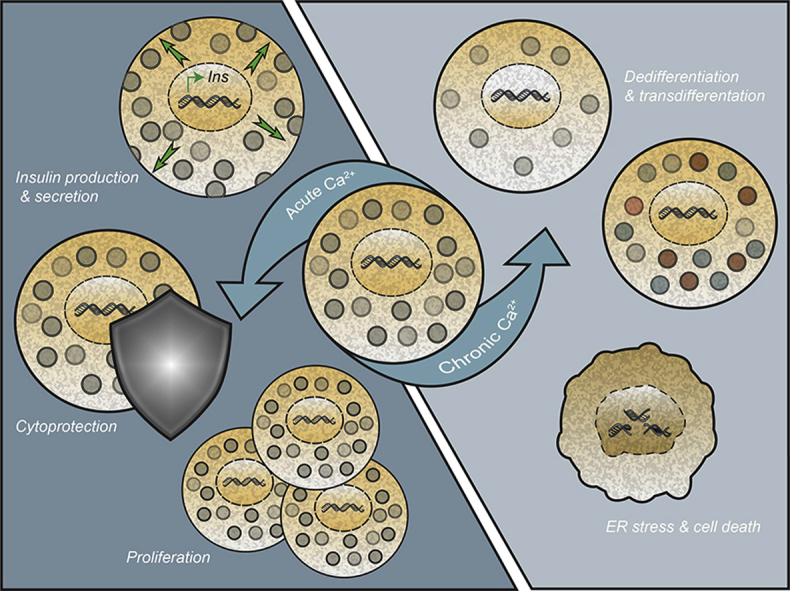
Acute increases in intracellular Ca 2+ stimulate a number of signaling cascades, resulting in (de-)phosphorylation events and activation of downstream transcription factors. The short-term stimulation of these Ca 2+ signaling pathways promotes numerous cellular processes critical to β-cell function, including increased viability, replication, and insulin production and secretion. Conversely, chronic stimulation of Ca 2+ signaling pathways increases β-cell ER stress and results in the loss of β-cell differentiation status. Together, decades of study demonstrate that Ca 2+ movement is tightly regulated within the β-cell, which is at least partially due to its dual roles as a potent signaling molecule.
Graphical abstract
Related collections
Most cited references133
- Record: found
- Abstract: found
- Article: not found
NCLX is an essential component of mitochondrial Na+/Ca2+ exchange.
- Record: found
- Abstract: found
- Article: not found
NAADP mobilizes calcium from acidic organelles through two-pore channels
- Record: found
- Abstract: found
- Article: not found