- Record: found
- Abstract: found
- Article: found
Multilevel Framework for Analysis of Protein Folding Involving Disulfide Bond Formation

Read this article at
Abstract
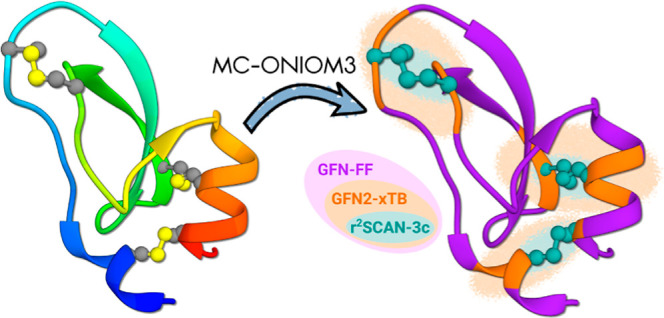
In this study, a three-layered multicenter ONIOM approach is implemented to characterize the naive folding pathway of bovine pancreatic trypsin inhibitor (BPTI). Each layer represents a distinct level of theory, where the initial layer, encompassing the entire protein, is modeled by a general all-atom force-field GFN-FF. An intermediate electronic structure layer consisting of three multicenter fragments is introduced with the state-of-the-art semiempirical tight-binding method GFN2- xTB. Higher accuracy, specifically addressing the breaking and formation of the three disulfide bonds, is achieved at the innermost layer using the composite DFT method r 2SCAN-3c. Our analysis sheds light on the structural stability of BPTI, particularly the significance of interlinking disulfide bonds. The accuracy and efficiency of the multicenter QM/SQM/MM approach are benchmarked using the oxidative formation of cystine. For the folding pathway of BPTI, relative stabilities are investigated through the calculation of free energy contributions for selected intermediates, focusing on the impact of the disulfide bond. Our results highlight the intricate trade-off between accuracy and computational cost, demonstrating that the multicenter ONIOM approach provides a well-balanced and comprehensive solution to describe electronic structure effects in biomolecular systems. We conclude that multiscale energy landscape exploration provides a robust methodology for the study of intriguing biological targets.
Related collections
Most cited references77

- Record: found
- Abstract: found
- Article: found
Highly accurate protein structure prediction with AlphaFold
- Record: found
- Abstract: found
- Article: not found
Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy.
- Record: found
- Abstract: not found
- Article: not found